
·综述
单细胞分选与测序技术在环境微生物领域的应用及前景
(南京农业大学资源与环境科学学院/江苏省固体有机废弃物资源化高新技术研究重点实验室,江苏 南京 210095)
Application and prospect of single-cell isolation and sequencing technology in environmental microbiology
(College of Resources and Environmental Sciences/Jiangsu Provincial Key Laboratory for Solid Organic Waste Utilization, Nanjing Agricultural University, Nanjing 210095, China)
single-cell isolation; single-cell sequencing; single-cell genomics; environmental microbiology
基金项目:国家自然科学基金项目(42307174)
*通信作者:徐琪程,博士,副研究员,主要从事农田土壤微生物研究,E-mail:qichengxu@njau.edu.cn。
-
随着对环境微生物生态系统认知的深化,对其深入研究的需求持续增加,传统的培养组学和环境DNA测序等研究手段已经难以满足对微生物物种和功能多样性的全面了解。单细胞分选与测序技术为环境微生物研究开创了全新的视角,成为该领域的热点与前沿。单细胞技术将微生物研究的焦点从群落水平深入到个体细胞层面,通过精准分选并测序单个微生物细胞,深入挖掘微生物的种间和种内多样性以及功能特性。本综述介绍单细胞技术在环境微生物研究中的应用,详细论述单细胞分选和基因组扩增的关键过程,并探讨单细胞测序数据解析过程中的生物信息学基本流程,为单细胞分选与测序技术在环境微生物研究中的发展提供借鉴和启示。
As our understanding of environmental microbial ecosystems deepened, the demand for in-depth studies in this area continued to grow. Traditional methods such as culturomics and environmental DNA sequencing were no longer sufficient to fully understand the species and functional diversity of microbes. The single-cell isolation and sequencing technology emerged as a new paradigm in environmental microbiology, representing a hot topic and cutting-edge advancement. This single-cell technology shifted the focus of microbial research from the community level to the individual cell level, allowing for the precise isolation and sequencing of single microbial cells. This approach provided a deeper understanding of the interspecies and intraspecies diversity and functional characteristics of microbes. This review introduced the application of single-cell technology in environmental microbiology, elaborated on the key processes of single-cell isolation and genome amplification, and discussed the bioinformatic flow of the single-cell sequencing data analysis. It aimed to provide insights and inspiration for the development of single-cell technology in environmental microbiology. 引言
在生态系统中,生物与生物之间以及生物与环境之间的相互作用发生在从分子到群落以及物种内部或物种之间等多个生态尺度上,并受到各种生物及非生物因素的影响。为了全面理解生态和进化过程,需要从不同生态尺度和角度出发,获取更加详尽的信息[1]。微生物个体(细胞)作为研究环境微生物的基本单位[2-3],是连接生态系统、群落和种群的纽带。在微生物代谢过程中,基因组和代谢途径的微小变化会改变微生物互作关系以及微生物群落构建过程,从而对生态系统产生重大影响。因此,研究大尺度上的生态机制需要关注微生物个体的作用过程,这也是当今环境微生物研究所面临的挑战之一[4]。
在微生物学领域,宏基因组学(metagenomics)、宏转录组学(metatranscriptomics)和培养组学(culturomics)是目前常用于研究微生物群落的方法,在微生物系统发育关系[5]、物种多样性和丰度[6]、代谢能力[7]以及功能多样性[8]等研究中发挥重要作用。然而,这些方法在描述微生物群落及其功能时存在一定的局限性:宏组学(meta-omics)无法准确提供微生物个体水平的信息[9]; 培养组学关注的微生物种群或数量非常有限,难以提供群落水平的生态信息,并且评估过程存在一定的主观性[10-11]。不同生态尺度下群落组成的不确定性和微生物间相互作用的复杂性[3]进一步增加了选择恰当研究手段的难度[12-13]。
单细胞技术的出现为环境微生物研究带来了新的突破。它能够在微生物基因组进行全面研究[14-16],准确获取每个细胞的基因组信息,从而高度代表原始个体[17]。通过单细胞技术,可以大量获取环境微生物基因组信息,注释功能并重建代谢途径,从而揭示细胞间基因表达和代谢途径的异质性。这在探索微生物多样性、种群遗传与进化,以及微生物相互作用等方面具有巨大潜力。此外,单细胞基因组学信息能够提供系统发育和功能性状之间的联系,揭示采样时单个细胞的生理状态[18]。结合细胞分选工具,如基于细胞图像的全自动单细胞分选仪(CellenONE),可以根据细胞的完整性、生理状态或功能标记来分选细胞,并最大限度地减少宿主或细胞外DNA的污染。因此,单细胞组学方法不仅能够补充宏组学研究在个体尺度上的不足,还能够从环境样本中挖掘培养组学难以解析的群落水平相互作用。
本综述重点介绍了在环境微生物领域中已应用或具有应用前景的单细胞分选技术和单细胞基因组测序技术,详细论述了这些技术在突破微生物学传统分离培养限制、探究微生物多样性、剖析种群遗传和进化,以及破译微生物间或微生物与宿主的互作等方面的应用,并对现存生物信息学方法在挖掘单细胞数据方面的潜力进行了展望,以期为单细胞分选与测序技术在环境微生物中的应用和发展提供理论依据。

图1 生态复杂性梯度、研究尺度和相关方法
Fig.1 Gradient of ecological complexity,study scales and associated approaches1 单细胞分选与测序技术在环境微生物中的应用和优势
1.1 突破传统分离培养限制
传统单细胞培养组学采用“先培养,后筛选”的策略,将细胞置于特定的培养条件下,直至形成单菌落,然后通过纯培养筛选特定的代谢表型组。然而,自然界中的可培养微生物仅占很小比例[19]。同时,可培养微生物还受到培养活性、条件及培养纯度等多重因素影响,导致分离效率低下[18]。单细胞分离培养法提供了一条新的途径,即“先分离,后培养”,这种策略理论上可以极大提高分离培养的通量和效率。此外,单细胞分离技术可以利用荧光标记、拉曼光谱等技术识别目标细胞,从而能够更有针对性地从接近原位的环境中分离出目标细胞,并进行后续扩繁培养。对于难以培养的微生物,单细胞分离技术能够与测序手段相结合,提供该微生物的代谢通路等信息。基于这些信息,有针对性地补充生长限制因子,有望实现这类微生物的可培养化。单细胞分离培养技术是以单个细胞为起点的多代的增殖。在此过程中,温和的细胞处理方式以及精确的细胞分离操作可以显著提升细胞培养的效率[20-22]。
1.2 探究微生物多样性
单细胞技术可以补充和验证宏组学手段在真菌[23]、细菌[24]等领域所揭示的物种多样性,通过单细胞方法获取的微生物序列集合中,可能含有全新的微生物分支。环境微生物群落往往由少数高丰度物种和大量低丰度物种共同构成[25]。宏基因组数据虽然能够反映环境中微生物的整体功能,但难以揭示不同微生物个体间的功能异质性和稀有物种的信息。尽管通过宏基因组分箱(binning)的生物信息学方法可以获得组装的基因组(metagenome-assembled genome,MAG),并注释功能信息,但此方法一般局限于环境中丰度较高的物种。此外,分箱过程无法直接确定属于某一微生物物种的所有序列,存在组装错误的风险。相比之下,单细胞组学以单个细胞为测序单位,能够最大限度地保证序列来源的可溯性,这有助于发掘宏组学研究中被大量基因片段所掩盖的稀有物种信息。鉴于稀有物种在环境扰动中对微生物群落的稳定性和功能的关键作用[26-27],单细胞技术捕捉稀有物种多样性与功能信息的能力对理解微生物群落响应环境扰动的机制尤为重要。
1.3 剖析种群遗传与进化
单细胞基因组学不仅有助于了解微生物的种类、代谢途径和功能信息,而且能够揭示种群遗传与进化的机制,这为探索众多生态学理论和假设提供了方法。单细胞测序技术可以通过捕捉个体间的基因组变异来剖析自然环境中的种群异质性[28],从而阐明微生物种内多样性、生态位划分,并在基因水平转移的研究中发挥重要作用[29-30]。例如,基于微生物单细胞基因组研究阐明短链脂肪酸、共生因子等合成机制和代谢途径[31-32],评估物种在不同生态位的代谢功能异质性[33]。此前,生态进化过程的研究主要依赖生态模型[34]或体外试验[35],而单细胞技术的出现为验证这些假设提供新的途径,并突出微生物群落遗传进化模式的重要性。
1.4 破译微生物间或微生物与宿主间的相互作用
尽管当前已对环境微生物群落的构成进行了广泛研究,但微生物间以及微生物与宿主间相互作用的机制仍不明晰[36]。单细胞组学可以获取细胞内全部遗传物质的信息,进而在细胞自身DNA和外来遗传信息之间建立联系[37],通过分析单细胞和细胞外遗传元件之间的水平基因转移等过程,揭示自然栖息地中微生物群内部以及微生物群与宿主之间的相互作用。Munson-McGee等[38]将单细胞基因组学与宏基因组学相结合,将在不同细胞中发现的病毒相互关联。基于单细胞基因组(single-amplified genome,SAG)分析,无需借助培养组学就能识别侵染单个宿主的病毒类型和单个病毒的宿主范围,增强对病毒-宿主相互作用的理解。在海洋细菌和古细菌研究中,通过单细胞技术鉴定出的多种次级代谢物合成途径为解释细胞间的相互作用提供了新思路[39]。单细胞基因组学可通过监测细菌种群基因的动态以验证细菌种群变化的假设[40],这对于了解微生物如何在群落内发生相互作用以及它们对生态系统的可能影响是至关重要的[41-42]。
单细胞组学提供了单个微生物的功能、代谢特征以及种群、群落水平的物种组成和整体功能信息,将不同生态尺度的结果信息连接起来,提供了更广泛的生态理解(图1)。此外,单细胞技术在环境微生物研究中还具有突破传统分离培养限制、探究微生物多样性、剖析种群遗传与进化、破译微生物间或微生物与宿主间的相互作用等优势。
2 单细胞分选与测序技术的关键步骤
2.1 单细胞分选
单细胞分离是单细胞技术流程的首要步骤,其准确性将直接影响后续的测序和结果分析。目前主流的分离技术包括有限稀释(limiting dilution)、显微操作(micromanipulator)、激光捕获显微切割(laser capture microdissection)、荧光流式细胞分选(fluorescence activated cell sorting)、单细胞拉曼光谱(single-cell raman-activated sorting)[44-45]以及微流控技术(microfluidics)[46]。
有限稀释法只需借助标准移液工具进行样本稀释,但由于细胞在等分液体中的分布遵循泊松分布,因此效率较低。根据泊松分布,只有约1/3的细胞培养板孔中能成功捕获单个细胞,且需要额外步骤,如显微镜观察,来确认哪些孔含有单细胞。显微操作技术则通过在高倍显微镜下使用玻璃毛细管或微型探针装置实现对微生物的提取、分离。然而,该技术对环境样本有要求,仅适用于液体样本分选,且依赖手动操作,对操作者的技能要求较高,分选通量低[47]。激光捕获显微切割是将样本冻存或封存在石蜡中后,放置于高倍显微镜下,先进行显色标记,再利用激光能量对目标单细胞进行切割,从而实现对细胞的特异性获取[48-49]。然而在分离过程中,激光能量可能会损害细胞的完整性,并且该方法成本较高。激光捕获显微切割在生物医学研究等领域应用较多,在环境微生物领域中常用于动植物宿主中病原微生物的分离[50]。荧光流式细胞分选技术通过荧光标记细胞,并利用细胞对荧光散射信号的特异性,实现对同类型细胞的精准分选[51]。该技术以细胞标志物为依据,具有高回收率和精度,目前已广泛应用于环境样本中细菌和丝状真菌的分选[52-23]。单细胞拉曼光谱技术通过检测代谢分子的拉曼光谱,反映物质分子内部化学键的状态,从而确定微生物的种类、生理特征、营养状况和突变表型等“生物指纹”[53],同时结合光学镊子实现对目标单细胞的分选[54]。这种方法具有非标记、无接触等优点,确保细胞完整性,该技术已实现对污水中具有强溶磷作用微生物的分选[54]。但光谱成像速度较慢、通量较低等缺点限制其推广应用。微流控技术是一种高通量的单细胞分离技术,液滴微流控技术通过包裹单细胞形成油滴,释放油滴后获得单个细胞基因组[55-56]。尽管液滴微流控技术具有操作简单、高通量和成本较低等优势,但存在无法识别空液滴和液滴含有多个细胞的缺点[46]。此外,由于液滴微流控技术优先保证细胞的通量与整体流程运行速度,其对于微生物的裂解并不充分,这对后续的基因组扩增和测序环节产生显著影响。
新兴微流控技术平台CellenONE为复杂环境中的单细胞生态学研究创造了全新的机遇[18]。单细胞分离与测序技术的主要步骤见图2。CellenONE平台具有高精度的单细胞分离与精密的皮升级试剂分配功能,配备可灭菌的玻璃毛细管用于样本吸入和分选,并通过电压和脉冲产生微液滴。相较于传统液滴微流控技术,CellenONE突破了无法识别液滴中细胞的局限,通过搭载高倍显微相机,实现对单个细胞的精准识别。当液滴为空或有多个细胞或碎片时,系统将自动舍弃该液滴。借助内置的高分辨率显微镜,CellenONE能够在毛细管尖端对单个细胞进行成像,并通过系统成像技术和算法,计算细胞的直径、伸长率、圆度、亮度和荧光强度等参数。通过对这些参数进行个性化设置,精确筛选出目标细胞[57]。此外,CellenONE配备2个可控温度的孔板支架,能够将单细胞或试剂分配至各种类型的微孔板(如96、384和1 536孔板),实现高通量、无监督和全自动的操作系统[58]。
已有研究成功将CellenONE平台应用于人体细胞分离[59]。在此基础上,CellenONE平台针对微生物特性开发了一款名为microLIFE的软件。该软件专用于微生物分选,其具备识别0.8 μm及以上微生物形态的能力,利用高分辨率显微摄像头获取的形态数据来判定和分选微生物。相比于类似的荧光细胞分选技术,CellenONE-microLIFE的分选通量相当,但是对于脆弱微生物或者特殊样本而言,它可以省略对样本进行荧光或同位素标记处理步骤,从而最大程度地减少对样本微生物的影响。这意味着该技术可以更加温和地处理微生物样本,同时保持高效的分选能力和微生物活性。
在土壤[60]、植物内生菌研究中,该技术可以成功分选出单个微生物并获得其基因组,为单细胞微生物生态研究带来广泛的前景。CellenONE平台通过其高精度的单细胞分离和精密试剂分配功能,结合高倍显微成像和自动化操作系统,为复杂环境中单细胞生态学的研究提供了强有力的技术支持。
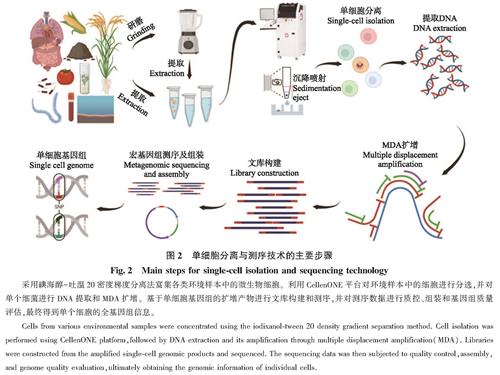
图2 单细胞分离与测序技术的主要步骤
Fig.2 Main steps for single-cell isolation and sequencing technology2.2 单细胞基因组扩增
单个细胞的DNA含量通常无法满足高通量测序的需求[43],因此需要将单细胞内的核酸扩增至10万倍以上[61-62]。单细胞扩增技术最理想的状态是将基因组完整、准确地扩增,为了满足这一需求,单细胞扩增技术不断革新。目前的方法包括简并寡核苷酸引物PCR(degenerate oligonucleotide-primed polymerase chain reaction,DOP-PCR)、多重置换扩增(multiple displacement amplification,MDA)以及多次退火环状循环扩增技术(multiple annealing and looping-based amplification cycles,MALBAC)[63]。
DOP-PCR利用带有ATGTGG的简并寡核苷酸作为引物,该引物的3'端在基因组DNA中高频出现,从而实现对细胞全基因组的扩增[64]。DOP-PCR具有普通PCR的指数扩增特性,由于扩增的起始位点由特异的6个碱基决定,退火过程具有序列倾向性,这可能导致基因组不同序列之间的扩增差异。此外,DOP-PCR扩增的基因组覆盖度较低,对于拷贝数变异(copy number variation,CNV)的检测效果也较差[65]。
目前,MDA是单细胞扩增的主流技术[66],采用等温链置换扩增法。MDA利用φ29 DNA聚合酶和随机六聚体实现整个基因组的扩增。φ29 DNA聚合酶具有3'到5'端的外切酶活性[67],能够在多个位点同时与模板DNA开始复制,从而迅速获得大量高分子质量的DNA[68]。MDA可对全基因组进行高保真均匀扩增,是覆盖度较高的全基因组扩增方法[14,69]。此外,MDA在大片段文库构建方面具有显著优势,能够提供更全面、准确和连续的基因组信息,也更适用于复杂的环境样本。然而,MDA仍属于指数扩增,存在PCR反应的序列偏好。尽管如此,Leung等[70]用129个正常二倍体细胞的大数据集检测液滴MDA(droplet MDA)技术的性能,结果显示其在扩增均匀性、基因组覆盖度和稳健性方面的表现均优于其他单细胞全基因组扩增方法,实现了高达80%的单细胞基因组覆盖率,并在靶向测序中有效检测单核苷酸变异(single nucleotide variant,SNV)。
MALBAC技术于2013年初次应用于胚胎遗传诊断,现已广泛用于人类单倍型研究[71]。其扩增引物的3'端包含8 bp的随机序列,而5'端包含27 bp的相同序列。MALBAC扩增过程中,随机引物首先与模板DNA结合并进行拟线性扩增。在此阶段,特定设计的引物在扩增末端形成独特的环状结构,使得这些扩增产物可被特异性识别。拟线性扩增后,采用常规的PCR方法对这些环状结构的扩增产物进行指数扩增[72]。相比于DOP-PCR和MDA,MALBAC的拟线性扩增有助于降低指数扩增的序列偏好性,提高CNV检测的准确度[73]。然而,由于MALBAC使用的聚合酶保真性较低且扩增循环数较高[74],导致其SNV的假阳性概率比MDA高出40倍[75-76]。这可能对单细胞基因组的完整性产生较大影响,因此在微生物单细胞研究中,MALBAC技术需要进一步改进。
在单细胞技术流程中,单细胞分离与基因组扩增是最为关键的步骤。在进行单细胞分离时,需要选择适当的技术平台,确保高通量的同时维持高回收率,预期在短时间内有效分离出大量细胞,并尽可能减少细胞损失。在分离过程中需要保护细胞的完整性与活性,以避免对后续培养组学试验造成干扰。此外,由于单个细胞中的DNA含量极低,通常仅为皮克级别,而构建基因组测序文库所需的下限为1 ng,在构建文库之前,需对单细胞的全基因组进行高效且均匀的扩增[43]。这一步骤对扩增方法提出了极高的要求,需要克服碱基错配、扩增偏好性等技术难题,以确保最终结果的准确性和可靠性。
3 单细胞测序的生物信息学分析流程
3.1 组装方法
与常规测序数据分析流程类似,单细胞测序数据首先需要进行质控。用Fastqc[77]、trimmomatic[78]和Bowtie2[79]工具对原始数据进行裁剪和过滤,去除低质量序列和接头序列,去除宿主基因片段等,获得高质量短序列(clean reads)。每个单细胞基因组数据可以根据建库时独有的标签序列(Barcode)进行区分,随后进行单个细胞样本组装,这种方法节省测序成本以及基因组组装所需的计算资源。由于高度不均匀的序列覆盖率、较高的测序错误率和嵌合体等问题,单细胞数据的组装具有一定挑战性。SPAdes工具在单细胞数据组装方面表现出色[80],在使用SPAdes短序列组装工具对clean reads进行组装时,通过多次调整kmer参数,可以获得最优质量的SAG[54]。为了获得细胞基因组的功能,使用Prokka[81]预测单细胞基因组的ORF(open reading frames),并使用KofamKOALA[82]和eggNOG-mapper[83]对ORF进行功能注释。Jing等[54]通过完整的单细胞分选、培养和测序流程成功在原位分离培养出具有溶解有机磷能力的微生物。这些微生物在原位条件下的溶磷活性相较于纯培养条件提高了90%~100%。同时该研究重建了相关单细胞基因组的代谢模型,揭示了微生物在污水中高效溶解有机磷的机制。
3.2 单细胞基因组质量评估
为了评估单细胞测序获得的SAG质量,通常使用checkM等工具来评估其完整度和污染度[84]。然而,研究发现单个细胞样本组装的SAG普遍存在完整度较低的问题[1]。为了提升SAG的完整度,一些研究进行了方法改进:使用具有更高基因组回收能力的φ29 DNA聚合酶[69]; 在单细胞裂解过程中,加入溶菌酶或提高裂解温度,以充分裂解[69]。此外,相较于单个单细胞样本的组装,将亲缘关系较近的单细胞样本混合后进行组装可以提高SAG的完整度[16]。这是因为不同细胞间存在共享的高度保守基因组区域,这些区域在组装过程中提供了额外的连接点,使得组装更为容易和完整。
单细胞测序通过研究单个细胞的基因组学信息,为了解复杂微生物群落提供了独特的视角。相较于宏基因组测序和扩增子测序,单细胞测序技术可以更准确地鉴定物种分类,揭示功能基因信息,并挖掘微生物种群的多样性与互作关系。
4 总结与展望
在环境中不可培养的微生物被称为“暗物质”,而单细胞分选与测序技术则是解锁“暗物质”的关键。本综述介绍单细胞相关技术的优缺点,强调根据样本类型、分选通量和后续试验需求来选择适合技术的重要性,并以CellenONE-microLIFE平台为例,概述单细胞分选与测序技术的主要方法步骤。
目前已发表的微生物单细胞研究中,所分离、测序的细胞数量有限,这种低通量的单细胞技术流程导致单细胞组学在代表性上受到诟病,其主要原因是微生物单细胞技术在试验、成本上仍存在挑战。例如:细胞裂解、扩增方面的挑战; 不同微生物细胞膜组成结构各异; 缺乏高效、高通量的裂解微生物单细胞的方法。目前大多单细胞研究中仍采用热、冻融、超声、酶等方式,需找寻一种对普遍微生物细胞膜能有效裂解的酶或者物理方法。虽然MDA扩增技术所获得的基因组已具有较高的完整度,但基因组的质量在一定程度上受到扩增均匀度影响,试验上可运用皮升或纳升级别反应体系抑制MDA偏倚,后续利用生物信息学工具识别排除污染物,从两方面抑制MDA的偏倚扩增减少污染。单细胞扩增和文库构建的费用占据单细胞试验的大部分成本,减少反应体积和借助全自动建库仪是降低成本、减少样本污染的可行性方法。此外,未来从单细胞基因组向单细胞转录组发展的过程中,存在微生物细胞膜裂解方法无法统一、微生物缺乏多聚腺苷酸化的mRNA以及微生物表达的转录本数量小等问题。
在具有低成本高回报率的高通量单细胞技术出现前,将单细胞组学与宏基因组学结合运用于同一项研究中是目前环境微生物研究的最优解; 宏基因组获得数据构建假设,通过单细胞组学进行检验,能更准确评估复杂微生物群中的微生物基因组多样性和基因组功能。单细胞组学所获得的微生物基因组和培养组学数据,已能很好地解决微生物、宿主互作和遗传与进化相关问题。总的来说,单细胞分选与测序技术对于深入探索微生物生态学、理解地球生态系统功能以及完善地球生物的系统发育树具有重大意义。
-
参考文献
- [1]Lloyd K G,Steen A D,Ladau J,et al. Phylogenetically novel uncultured microbial cells dominate earth microbiomes[J]. mSystems,2018,3(5):e00055-18.
- [2]Coyte K Z,Rakoff-Nahoum S. Understanding competition and cooperation within the mammalian gut microbiome[J]. Current Biology,2019,29(11):R538-R544.
- [3]Pacheco A R,Segrè D. A multidimensional perspective on microbial interactions[J]. FEMS Microbiology Letters,2019,366(11):fnz125.
- [4]Dini-Andreote F,Kowalchuk G A,Prosser J I,et al. Towards meaningful scales in ecosystem microbiome research[J]. Environmental Microbiology,2021,23(1):1-4.
- [5]Alteio L V,Schulz F,Seshadri R,et al. Complementary metagenomic approaches improve reconstruction of microbial diversity in a forest soil[J]. mSystems,2020,5(2):e00768-19.
- [6]Daims H,Lebedeva E V,Pjevac P,et al. Complete nitrification by Nitrospira bacteria[J]. Nature,2015,528:504-509.
- [7]Cernava T,Erlacher A,Aschenbrenner I A,et al. Deciphering functional diversification within the lichen microbiota by meta-omics[J]. Microbiome,2017,5(1):82.
- [8]Parks D H,Rinke C,Chuvochina M,et al. Author correction:recovery of nearly 8 000 metagenome-assembled genomes substantially expands the tree of life[J]. Nature Microbiology,2018,3:253.
- [9]Hug L A,Baker B J,Anantharaman K,et al. A new view of the tree of life[J]. Nature Microbiology,2016,1:16048.
- [10]Li L L,Deng X T,Mee E T,et al. Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent[J]. Journal of Virological Methods,2015,213:139-146.
- [11]Mande S S,Mohammed M H,Ghosh T S. Classification of metagenomic sequences:methods and challenges[J]. Briefings in Bioinformatics,2012,13(6):669-681.
- [12]Werner G D A,Kiers E T. Partner selection in the mycorrhizal mutualism[J]. The New Phytologist,2015,205(4):1437-1442.
- [13]Amin S A,Hmelo L R,van Tol H M,et al. Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria[J]. Nature,2015,522:98-101.
- [14]Gawad C,Koh W,Quake S R. Single-cell genome sequencing:current state of the science[J]. Nature Reviews Genetics,2016,17:175-188.
- [15]Ishii S,Tago K,Senoo K. Single-cell analysis and isolation for microbiology and biotechnology:methods and applications[J]. Applied Microbiology and Biotechnology,2010,86(5):1281-1292.
- [16]Woyke T,Doud D F R,Schulz F. The trajectory of microbial single-cell sequencing[J]. Nature Methods,2017,14:1045-1054.
- [17]Dubey R K,Tripathi V,Prabha R,et al. Single-cell genomics and metagenomics for microbial diversity analysis[M]//Unravelling the Soil Microbiome. Cham:Springer,2020:33-49.
- [18]Mauger S,Monard C,Thion C,et al. Contribution of single-cell omics to microbial ecology[J]. Trends in Ecology & Evolution,2022,37(1):67-78.
- [19]Bodor A,Bounedjoum N,Vincze G E,et al. Challenges of unculturable bacteria:environmental perspectives[J]. Reviews in Environmental Science and BioTechnology,2020,19(1):1-22.
- [20]Rao C V,Wolf D M,Arkin A P. Control,exploitation and tolerance of intracellular noise[J]. Nature,2002,420:231-237.
- [21]Gracz A D,Williamson I A,Roche K C,et al. A high-throughput platform for stem cell niche co-cultures and downstream gene expression analysis[J]. Nature Cell Biology,2015,17:340-349.
- [22]Pushkarsky I,Tseng P,Black D,et al. Elastomeric sensor surfaces for high-throughput single-cell force cytometry[J]. Nature Biomedical Engineering,2018,2:124-137.
- [23]Bleichrodt R J,Read N D. Flow cytometry and FACS applied to filamentous fungi[J]. Fungal Biology Reviews,2019,33(1):1-15.
- [24]Marcy Y,Ouverney C,Bik E M,et al. Dissecting biological “dark matter”with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth[J]. Proc Natl Acad Sci USA,2007,104(29):11889-11894.
- [25]Sogin M L,Morrison H G,Huber J A,et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”[J]. Proc Natl Acad Sci USA,2006,103(32):12115-12120.
- [26]Shade A,Jones S E,Caporaso J G,et al. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity[J]. mBio,2014,5(4):e01371-14.
- [27]Xu Q C,Ling N,Quaiser A,et al. Rare bacteria assembly in soils is mainly driven by deterministic processes[J]. Microbial Ecology,2022,83(1):137-150.
- [28]Mas A,Lagadeuc Y,Vandenkoornhuyse P. Reflections on the predictability of evolution:toward a conceptual framework[J]. iScience,2020,23(11):101736.
- [29]Mas A,Jamshidi S,Lagadeuc Y,et al. Beyond the black queen hypothesis[J]. The ISME Journal,2016,10(9):2085-2091.
- [30]Zelezniak A,Andrejev S,Ponomarova O,et al. Metabolic dependencies drive species co-occurrence in diverse microbial communities[J]. Proc Natl Acad Sci USA,2015,112(20):6449-6454.
- [31]Chijiiwa R,Hosokawa M,Kogawa M,et al. Single-cell genomics of uncultured bacteria reveals dietary fiber responders in the mouse gut microbiota[J]. Microbiome,2020,8(1):5.
- [32]Siegl A,Kamke J,Hochmuth T,et al. Single-cell genomics reveals the lifestyle of Poribacteria,a candidate Phylum symbiotically associated with marine sponges[J]. The ISME Journal,2011,5(1):61-70.
- [33]Nunan N,Schmidt H,Raynaud X. The ecology of heterogeneity:soil bacterial communities and C dynamics[J]. Philosophical Transactions of the Royal Society of London Series B:Biological Sciences,2020,375(1798):20190249.
- [34]Wang M X,Liu X N,Nie Y,et al. Selfishness driving reductive evolution shapes interdependent patterns in spatially structured microbial communities[J]. The ISME Journal,2021,15(5):1387-1401.
- [35]Hennon G M M,Morris J J,Haley S T,et al. The impact of elevated CO2 on Prochlorococcus and microbial interactions with‘helper'bacterium Alteromonas[J]. The ISME Journal,2018,12(2):520-531.
- [36]Hassani M A,Durán P,Hacquard S. Microbial interactions within the plant holobiont[J]. Microbiome,2018,6(1):58.
- [37]Stepanauskas R. Wiretapping into microbial interactions by single cell genomics[J]. Frontiers in Microbiology,2015,6:258.
- [38]Munson-McGee J H,Peng S Y,Dewerff S,et al. A virus or more in(nearly)every cell:ubiquitous networks of virus-host interactions in extreme environments[J]. The ISME Journal,2018,12:1706-1714.
- [39]Pachiadaki M G,Brown J M,Brown J,et al. Charting the complexity of the marine microbiome through single-cell genomics[J]. Cell,2019,179(7):1623-1635.
- [40]Hatzenpichler R,Krukenberg V,Spietz R L,et al. Next-generation physiology approaches to study microbiome function at single cell level[J]. Nature Reviews Microbiology,2020,18:241-256.
- [41]Engel P,Stepanauskas R,Moran N A. Hidden diversity in honey bee gut symbionts detected by single-cell genomics[J]. PLoS Genetics,2014,10(9):e1004596.
- [42]McNulty R,Sritharan D,Liu S C,et al. Droplet-based single cell RNA sequencing of bacteria identifies known and previously unseen cellular states[J]. bioRxiv,2021. DOI:10.1101/2021.03.10.434868.
- [43]de Bourcy C F A,de Vlaminck I,Kanbar J N,et al. A quantitative comparison of single-cell whole genome amplification methods[J]. PLoS One,2014,9(8):e105585.
- [44]Li H Z,Yang K,Liao H,et al. Active antibiotic resistome in soils unraveled by single-cell isotope probing and targeted metagenomics[J]. Proc Natl Acad Sci USA,2022,119(40):e2201473119.
- [45]Yang K,Xu F,Zhu L J,et al. An isotope-labeled single-cell Raman spectroscopy approach for tracking the physiological evolution trajectory of bacteria toward antibiotic resistance[J]. Angewandte Chemie,2023,62(14):e202217412.
- [46]Zheng W S,Zhao S J,Yin Y H,et al. High-throughput,single-microbe genomics with strain resolution,applied to a human gut microbiome[J]. Science,2022,376(6597):eabm1483.
- [47]Ishøy T,Kvist T,Westermann P,et al. An improved method for single cell isolation of prokaryotes from meso-,thermo- and hyperthermophilic environments using micromanipulation[J]. Applied Microbiology and Biotechnology,2006,69(5):510-514.
- [48]Feng Y T,Wang S Y,Liu X Y,et al. Geometric constraint-triggered collagen expression mediates bacterial-host adhesion[J]. Nature Communications,2023,14:8165.
- [49]Guo W B,Hu Y N,Qian J Y,et al. Laser capture microdissection for biomedical research:towards high-throughput,multi-omics,and single-cell resolution[J]. Journal of Genetics and Genomics,2023,50(9):641-651.
- [50]Ramsay K,Jones M G K,Wang Z H. Laser capture microdissection:a novel approach to microanalysis of plant-microbe interactions[J]. Molecular Plant Pathology,2006,7(5):429-435.
- [51]Pereira H,Schulze P S C,Schüler L M,et al. Fluorescence activated cell-sorting principles and applications in microalgal biotechnology[J]. Algal Research,2018,30:113-120.
- [52]Espina L. An approach to increase the success rate of cultivation of soil bacteria based on fluorescence-activated cell sorting[J]. PLoS One,2020,15(8):e0237748.
- [53]曾琦,刘瑞,王楠,等. 拉曼光谱技术在医学检验领域中的研究进展(特邀)[J]. 光子学报,2021,50(10):1017002.
- [54]Jing X Y,Gong Y H,Pan H H,et al. Single-cell Raman-activated sorting and cultivation(scRACS-Culture)for assessing and mining in situ phosphate-solubilizing microbes from nature[J]. ISME Communications,2022,2:106.
- [55]Hsieh K,Mach K E,Zhang P F,et al. Combating antimicrobial resistance via single-cell diagnostic technologies powered by droplet microfluidics[J]. Accounts of Chemical Research,2022,55(2):123-133.
- [56]Anggraini D,Ota N,Shen Y G,et al. Recent advances in microfluidic devices for single-cell cultivation:methods and applications[J]. Lab on a Chip,2022,22(8):1438-1468.
- [57]Tejwani V,Chaudhari M,Rai T,et al. High-throughput and automation advances for accelerating single-cell cloning,monoclonality and early phase clone screening steps in mammalian cell line development for biologics production[J]. Biotechnology Progress,2021,37(6):e3208.
- [58]Liu Y C,Ansaryan S,Li X K,et al. Real-time monitoring of single-cell secretion with a high-throughput nanoplasmonic microarray[J]. Biosensors & Bioelectronics,2022,202:113955.
- [59]Vallone V F,Telugu N S,Fischer I,et al. Methods for automated single cell isolation and sub-cloning of human pluripotent stem cells[J]. Current Protocols in Stem Cell Biology,2020,55(1):e123.
- [60]Xu Q,Zhang H,Vandenkoornhuyse P,et al. Carbon starvation raises capacities in bacterial antibiotic resistance and viral auxiliary carbon metabolism in soils[J]. Proc Natl Acad Sci USA,2024,121:e2318160121.
- [61]Nyaku S T,Sripathi V R,Lawrence K,et al. Characterizing repeats in two whole-genome amplification methods in the reniform nematode genome[J]. International Journal of Genomics,2021,2021:5532885.
- [62]Cheung V G,Nelson S F. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA[J]. Proc Natl Acad Sci USA,1996,93(25):14676-14679.
- [63]王丹蕊,沈文丽,魏子艳,等. 单细胞测序技术在微生物生态领域中的应用[J]. 生物技术通报,2020,36(10):237-246.
- [64]Arneson N,Hughes S,Houlston R,et al. Whole-genome amplification by degenerate oligonucleotide primed PCR(DOP-PCR)[J]. CSH Protocols,2008,2008:pdb.prot4919.
- [65]Telenius H,Carter N P,Bebb C E,et al. Degenerate oligonucleotide-primed PCR:general amplification of target DNA by a single degenerate primer[J]. Genomics,1992,13(3):718-725.
- [66]Lizardi P M,Huang X,Zhu Z,et al. Mutation detection and single-molecule counting using isothermal rolling-circle amplification[J]. Nature Genetics,1998,19(3):225-232.
- [67]Dean F B,Nelson J R,Giesler T L,et al. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification[J]. Genome Research,2001,11(6):1095-1099.
- [68]Povilaitis T,Alzbutas G,Sukackaite R,et al. In vitro evolution of phi29 DNA polymerase using isothermal compartmentalized self replication technique[J]. Protein Engineering,Design and Selection,2016,29(12):617-628.
- [69]Donmez N,Brudno M. Hapsembler:an assembler for highly polymorphic genomes[M]//Lecture Notes in Computer Science. Heidelberg:Springer,2011:38-52.
- [70]Leung K,Klaus A,Lin B K,et al. Robust high-performance nanoliter-volume single-cell multiple displacement amplification on planar substrates[J]. Proc Natl Acad Sci USA,2016,113(30):8484-8489.
- [71]Huang J,Yan L Y,Fan W,et al. Validation of multiple annealing and looping-based amplification cycle sequencing for 24-chromosome aneuploidy screening of cleavage-stage embryos[J]. Fertility and Sterility,2014,102(6):1685-1691.
- [72]Huang L,Ma F,Chapman A,et al. Single-cell whole-genome amplification and sequencing:methodology and applications[J]. Annual Review of Genomics and Human Genetics,2015,16:79-102.
- [73]Baslan T,Hicks J. Single cell sequencing approaches for complex biological systems[J]. Current Opinion in Genetics & Development,2014,26:59-65.
- [74]Zhou X X,Xu Y,Zhu L B,et al. Comparison of multiple displacement amplification(MDA)and multiple annealing and looping-based amplification cycles(MALBAC)in limited DNA sequencing based on tube and droplet[J]. Micromachines,2020,11(7):645.
- [75]潘孝明,梁兴国. 全基因组扩增技术原理及研究进展[J]. 生物技术通报,2014(12):47-54.
- [76]Liu W Q,Zhang H M,Hu D,et al. The performance of MALBAC and MDA methods in the identification of concurrent mutations and aneuploidy screening to diagnose beta-thalassaemia disorders at the single-and multiple-cell levels[J]. Journal of Clinical Laboratory Analysis,2018,32(2):e22267.
- [77]Chen S F,Zhou Y Q,Chen Y R,et al. Fastp:an ultra-fast all-in-one FASTQ preprocessor[J]. Bioinformatics,2018,34(17):i884-i890.
- [78]Bolger A M,Lohse M,Usadel B. Trimmomatic:a flexible trimmer for Illumina sequence data[J]. Bioinformatics,2014,30(15):2114-2120.
- [79]Langmead B,Salzberg S L. Fast gapped-read alignment with Bowtie 2[J]. Nature Methods,2012,9:357-359.
- [80]Bankevich A,Nurk S,Antipov D,et al. SPAdes:a new genome assembly algorithm and its applications to single-cell sequencing[J]. Journal of Computational Biology,2012,19(5):455-477.
- [81]Seemann T. Prokka:rapid prokaryotic genome annotation[J]. Bioinformatics,2014,30(14):2068-2069.
- [82]Aramaki T,Blanc-Mathieu R,Endo H,et al. KofamKOALA:KEGG ortholog assignment based on profile HMM and adaptive score threshold[J]. Bioinformatics,2020,36(7):2251-2252.
- [83]Cantalapiedra C P,Hernández-Plaza A,Letunic I,et al. eggNOG-mapper v2:functional annotation,orthology assignments,and domain prediction at the metagenomic scale[J]. Molecular Biology and Evolution,2021,38(12):5825-5829.
- [1]Lloyd K G,Steen A D,Ladau J,et al. Phylogenetically novel uncultured microbial cells dominate earth microbiomes[J]. mSystems,2018,3(5):e00055-18.
南京农业大学学报